Warning: package 'ggplot2' was built under R version 4.4.3
t.test(Bodyweight ~ Diet, Weights_long)
Welch Two Sample t-test
data: Bodyweight by Diet
t = -2.7058, df = 17.892, p-value = 0.01453
alternative hypothesis: true difference in means between group chow and group fat is not equal to 0
95 percent confidence interval:
-7.1178367 -0.8941633
sample estimates:
mean in group chow mean in group fat
25.603 29.609
Linear model
#grafify linear modelsimple_model(Weights_long,"Bodyweight","Diet")
#anova tableanova(lm(Bodyweight ~ Diet, data = Weights_long))
Analysis of Variance Table
Response: Bodyweight
Df Sum Sq Mean Sq F value Pr(>F)
Diet 1 80.24 80.24 7.3212 0.01447 *
Residuals 18 197.28 10.96
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
You can get more information on the linear model with the summary command. To be able to run this, and other diagnostics, you must first save the model as an object in your environment.
#save the model#grafify linear modellmod1 <-simple_model(Weights_long,"Bodyweight","Diet")#get detailed summarysummary(lmod1)
Call:
lm(formula = Bodyweight ~ Diet, data = Weights_long)
Residuals:
Min 1Q Median 3Q Max
-5.243 -1.667 -0.694 1.538 6.417
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 25.603 1.047 24.456 2.92e-15 ***
Dietfat 4.006 1.481 2.706 0.0145 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 3.311 on 18 degrees of freedom
Multiple R-squared: 0.2891, Adjusted R-squared: 0.2496
F-statistic: 7.321 on 1 and 18 DF, p-value: 0.01447
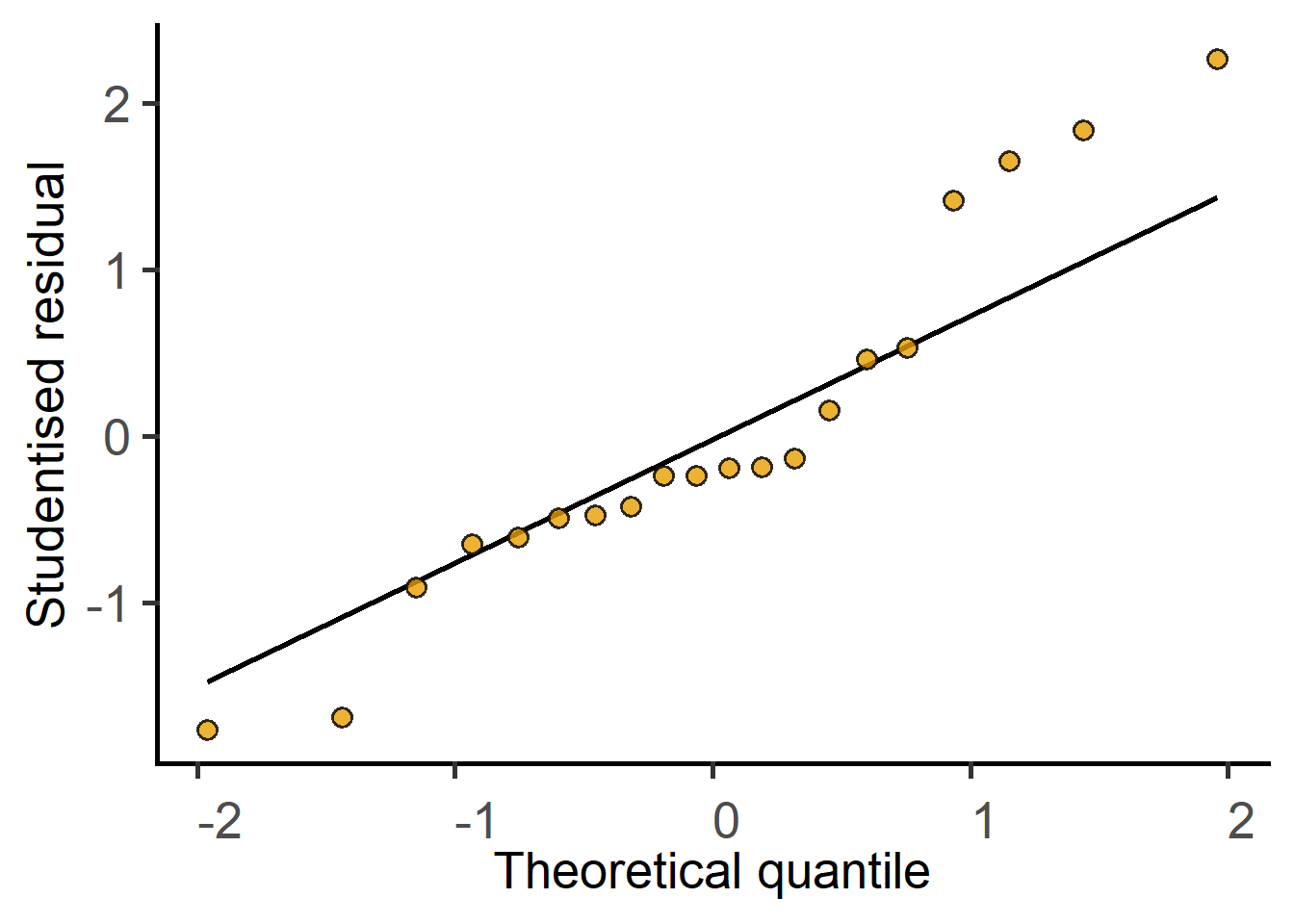
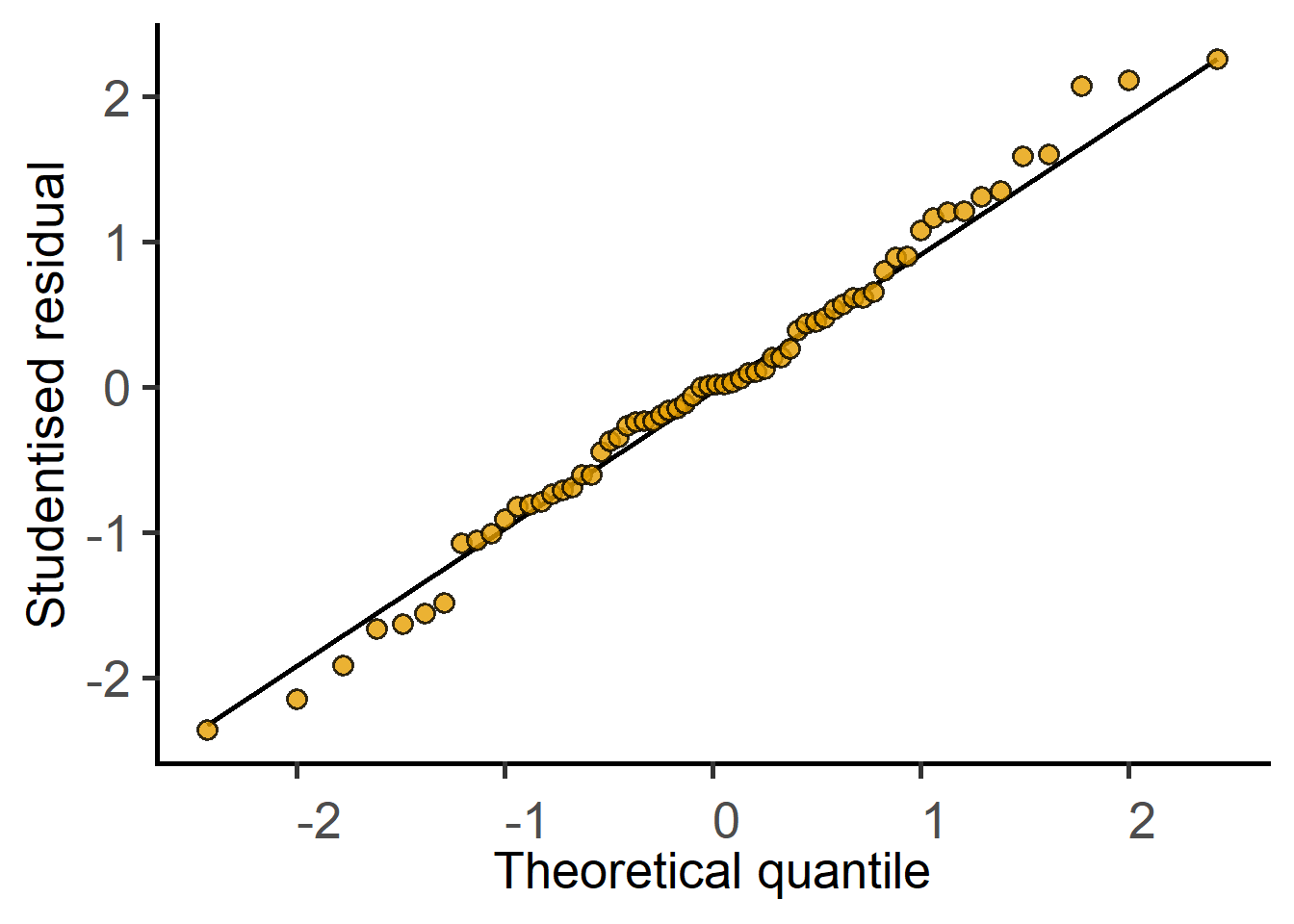
#get a residuals plotplot_qqmodel(lmod1)
Diagnosing a linear model with a plot of residuals
The residuals of the model fit should be assessed after the test. For a test result to be reliable, the residuals should be normally (or approximately normally) distributed.
Here we will use the data_t_pratio table.
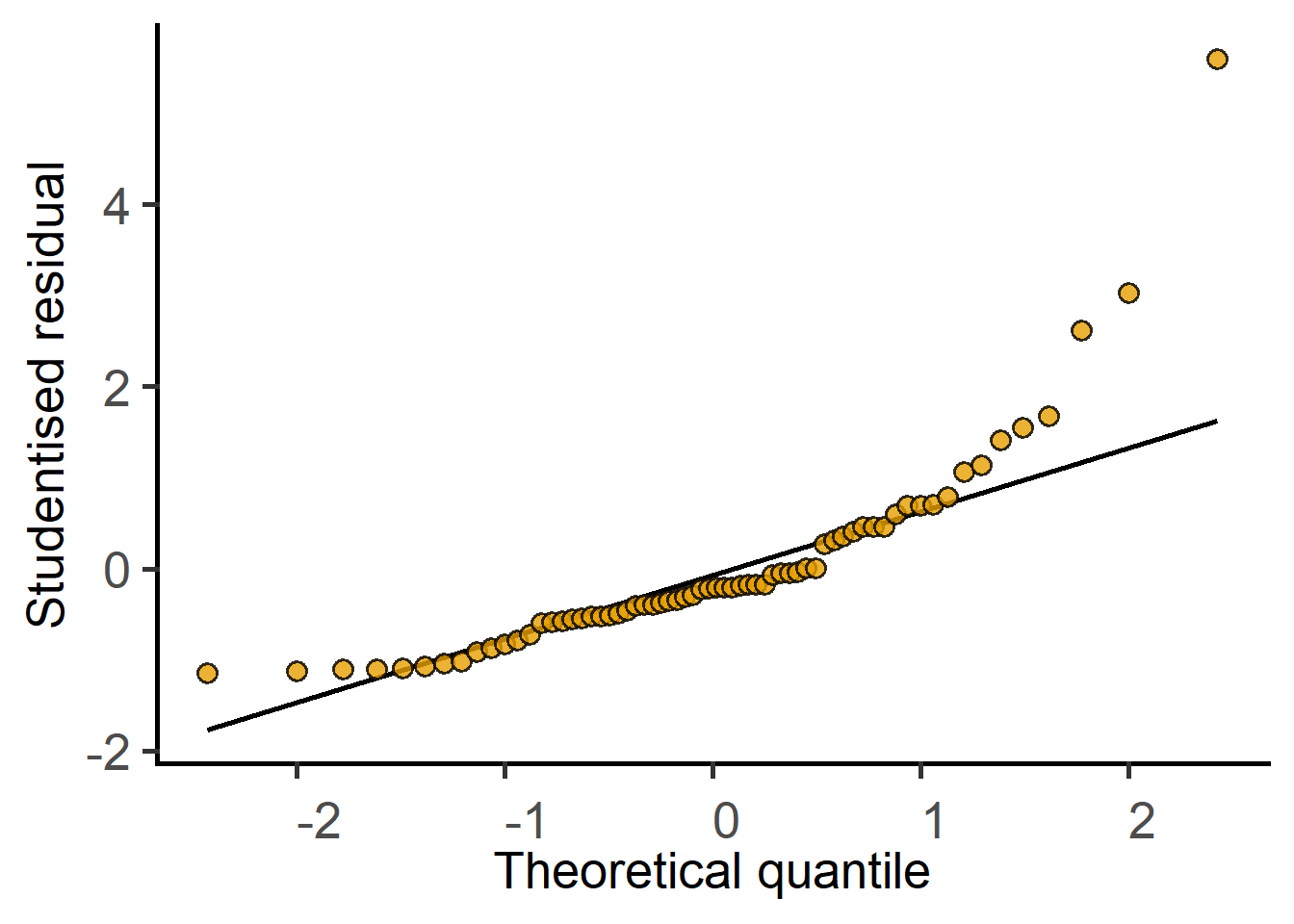
#fit model 1lin.m1 <-simple_model(data = data_t_pratio,Y_value ="Cytokine",Fixed_Factor ="Genotype")#fit a model of log-transformed datalin.m2 <-simple_model(data = data_t_pratio,Y_value ="log(Cytokine)",Fixed_Factor ="Genotype")#plot residuals of both modelsplot_qqmodel(lin.m1) #original data
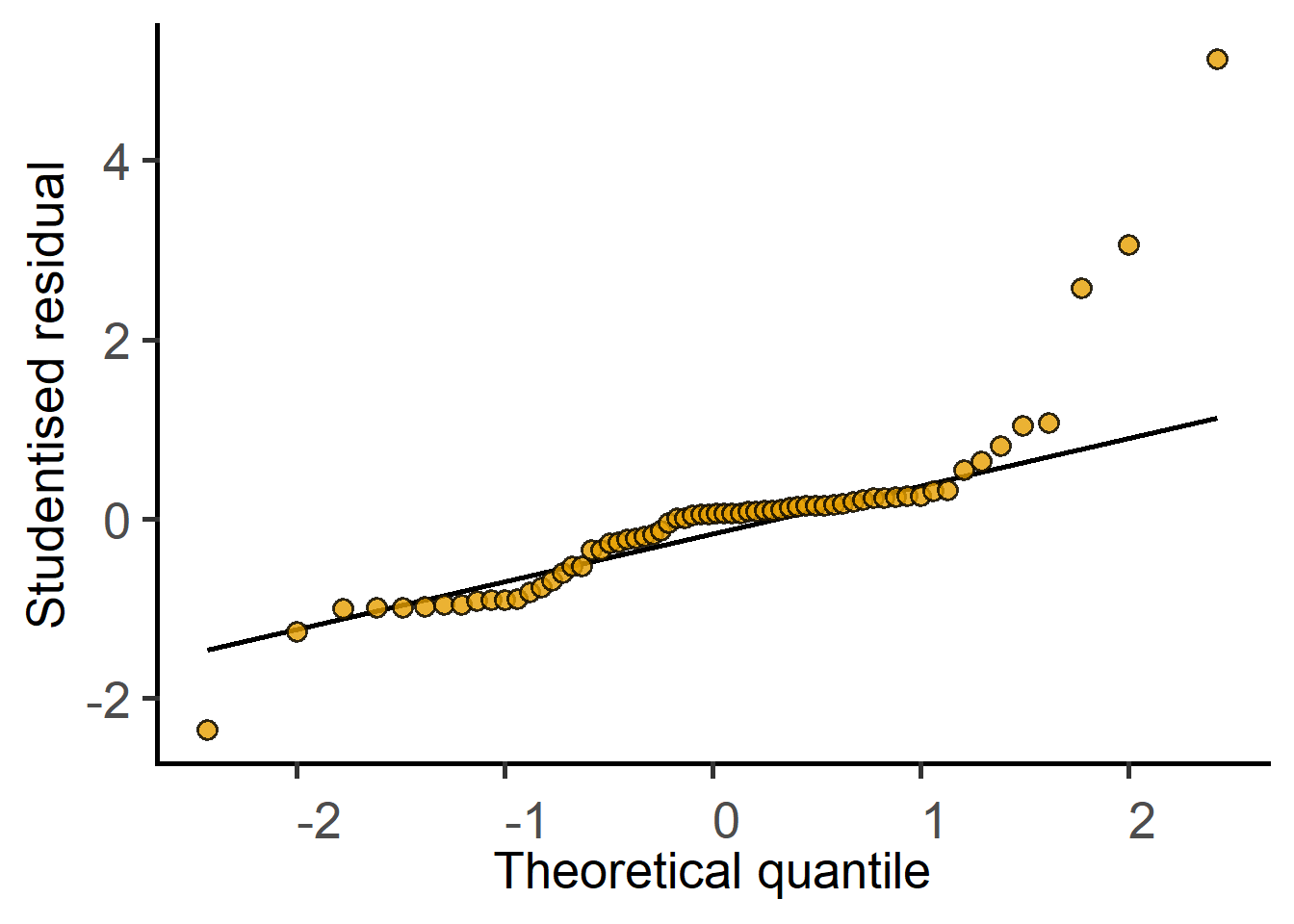
plot_qqmodel(lin.m2) #log-transformed data
Mixed effects models
Here, our data are matched by Experiment. We can use mixed effects models with Experiment as a random factor.
#fit mixed model 1mx.lin.m1 <-mixed_model(data = data_t_pratio,Y_value ="Cytokine",Fixed_Factor ="Genotype",Random_Factor ="Experiment")
The new argument `AvgRF` is set to TRUE by default in >=5.0.0). Response variable is averaged over levels of Fixed and Random factors. Use help for details.
#fit a model of log-transformed datamx.lin.m2 <-mixed_model(data = data_t_pratio,Y_value ="log(Cytokine)",Fixed_Factor ="Genotype",Random_Factor ="Experiment")
The new argument `AvgRF` is set to TRUE by default in >=5.0.0). Response variable is averaged over levels of Fixed and Random factors. Use help for details.
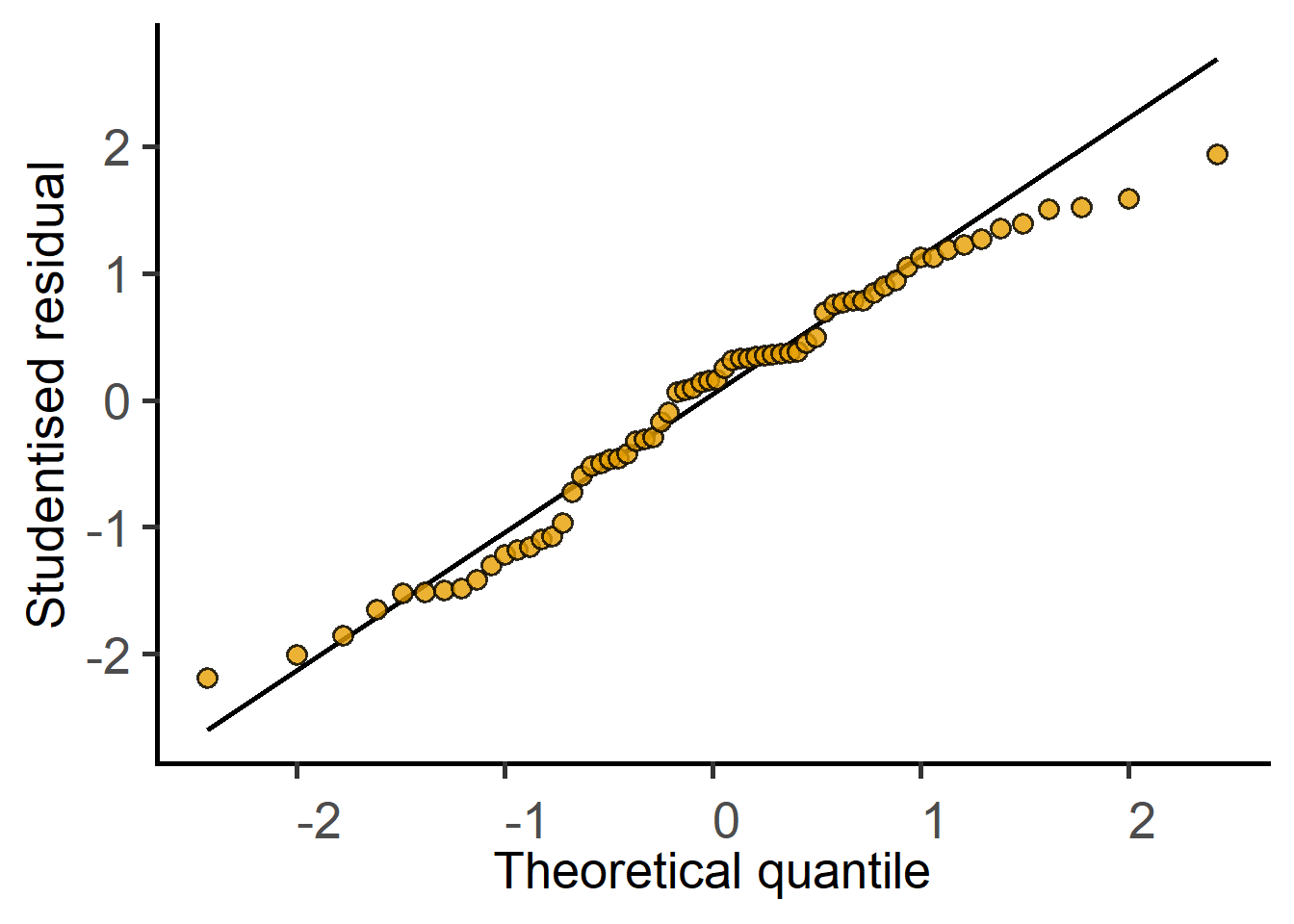
#plot residuals of both modelsplot_qqmodel(mx.lin.m1) #original data
plot_qqmodel(mx.lin.m2) #log-transformed data
ANOVA tables
In grafify, you can use very similar code to get ANOVA tables as for fitting a model. Once you diagnose which model fits the data better, use that one to get the ANOVA table and post-hoc comparisons.
The new argument `AvgRF` is set to TRUE by default in >=5.0.0). Response variable is averaged over levels of Fixed and Random factors. Use help for details.
Type II Analysis of Variance Table with Kenward-Roger's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
Genotype 6.4867 6.4867 1 32 68.873 1.758e-09 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
In this case (balanced designed of matched data without any missing data), a paired t test will give us a similar result. This can only be done with wide tables after an update to t.test.
#on original data#make a wide table first#use tidyrlibrary(tidyr)wdata_t_pratio <-pivot_wider(data_t_pratio, values_from = Cytokine,names_from = Genotype)t.test(wdata_t_pratio$WT, wdata_t_pratio$KO,paired =TRUE)
Paired t-test
data: wdata_t_pratio$WT and wdata_t_pratio$KO
t = -3.7385, df = 32, p-value = 0.0007256
alternative hypothesis: true mean difference is not equal to 0
95 percent confidence interval:
-4.523562 -1.332764
sample estimates:
mean difference
-2.928163
Paired t-test
data: log(wdata_t_pratio$WT) and log(wdata_t_pratio$KO)
t = -8.299, df = 32, p-value = 1.758e-09
alternative hypothesis: true mean difference is not equal to 0
95 percent confidence interval:
-0.7808986 -0.4731092
sample estimates:
mean difference
-0.6270039